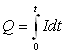Разработка методики определения ультрамикрограммовых количеств тяжелых металлов методом инверсионной вольтамперометрии
Глава 1. Анализ следовых количеств веществ и электрохимические инверсионные методы
1.1 Анализ следовых количеств веществ и проблемы, стоящие перед соответствующими методами анализа
1.2 Электрохимические методы анализа следовых количеств веществ
1.3 Основы электрохимических инверсионных методов
1.4 Реакции, используемые для электролитического накопления
1.5 Типы рабочих электродов
1.6 Методы исследования процесса растворения
1.7 Избирательность определения
1.8 Роль предварительного отделения в инверсионных электрохимических определениях
1.9 Состояние и перспективы метода
1.10 Примеры практических приложений инверсионных методов
Глава II. Методы исследования и методика проведения эксперимента
2.1 Инверсионная вольтамперометрия
2.1.1 Как можно сконцентрировать определяемый микроэлемент на индикаторном электроде
2.2 Методика выполнения измерений массовой концентрации ионов кадмия, свинца, меди и цинка в питьевых, природных и очищенных сточных водах методом инверсионной вольтамперометрии
2.2.1 Назначение и область применения методики
2.2.2 Характеристика погрешности измерений
2.2.3 Метод измерений
2.2.4 Средства измерений, вспомогательные устройства, реактивы и материалы
2.2.5 Условия безопасного проведения работ
2.2.6 Условия выполнения измерений
2.2.7 Подготовка к выполнению измерений
2.2.8 Выполнение измерений
2.2.9 Форма представления результатов измерений
2.2.10 Контроль характеристик погрешности
2.3. Оборудование, применяемое в работе
Глава III. Экспериментальная часть
3.1 Порядок работы
3.2 Электрохимические параметры выполнения измерений на СУ-электроде
3.3 Выполнение измерений на углеродном электроде
3.3.1 Регистрация вольтамперограммы фонового раствора
3.3.2 Регистрация вольтамперограмм анализируемого раствора пробы с добавкой стандартного раствора ионов тяжелых металлов
3.4 Обсуждение результатов
Выводы
Список литературы
Приложения
Актуальность. Современный уровень развития технологии, биологии, медицины, охраны окружающей среды и других областей науки и техники выдвигает задачу определения малых количеств веществ во все более сложных объектах; поэтому требования, предъявляемые к методам анализа следовых количеств веществ, постоянно повышаются. Наряду с другими методами при анализе следовых количеств широко применяются электрохимические инверсионные методы, поскольку для очень многих элементов при относительно простом аппаратурном оформлении они приводят к хорошо воспроизводимым и правильным результатам.
В последние годы число публикаций, посвященных инверсионным методам, неуклонно растет; это связано с появлением новых приборов, а также с переходом к использованию ртутных пленочных и твердых электродов.
В опубликованных до сих пор обзорных работах и книгах по электрохимическому инверсионному анализу основное внимание уделялось работам с классическим ртутным электродом. Представляет определенный интерес рассмотреть возможность использования пленочных и твердых электродов.
В нашей работе мы постарались представить краткий обзор исследований с использованием методов инверсионного анализа в анализе объектов окружающей среды.
Таким образом, целью данной работы явилась разработка методики определения ультрамикрограммовых количеств тяжелых металлов методом инверсионной вольтамперометрии.
Реализация выдвинутой цели предопределила решение ряда частных задач:
1. анализ литературы по проблеме анализа микрограммовых количеств тяжелых металлов методом инверсионной вольтамперометрии;
2. анализ методических разработок по анализу различных объектов окружающей среды методом инверсионной вольтамперометрии;
3. выработка методики определения следовых количеств тяжелых металлов методом инверсионной вольтамперометрии, исходя из имеющихся материалов и аппаратуры;
4. апробирование данной методики на водных объектах;
5. анализ полученных данных.
Глава 1. Анализ следовых количеств веществ и электрохимические инверсионные методы
1.1 Анализ следовых количеств веществ и проблемы, стоящие перед соответствующими методами анализа
Для успешного анализа следовых количеств веществ необходимо решить три проблемы:
1) достаточно сильно снизить предел обнаружения, т. е. повысить величину отношения сигнал/шум (например, величину отношения электрического тока к остаточному);
2) достигнуть требуемой избирательности, т. е. возможности определять следовые количества элементов на фоне других, присутствующих в концентрациях, на несколько порядков более высоких; эту проблему обычно невозможно решить без использования предварительного отделения;
3) приготовить химические реактивы требуемой степени чистоты и усовершенствовать технику работы с очень разбавленными растворами, содержание растворенного вещества в которых уменьшается из-за адсорбции на стенках ячейки, гидролиза и т. д.
При определении очень малых количеств веществ наиболее часто используются радиохимические методы, особенно активационный анализ и методы радиоактивных индикаторов. Практически эти методы характеризуются наиболее высокой чувствительностью среди всех методов, применяемых при анализе следовых количеств; так, достигаемый в оптимальных случаях предел обнаружения равен ~10-21 г, что соответствует приблизительно десяти атомам или молекулам. Из физико-химических методов анализа к радиометрическим методам по пределам обнаружения приближаются флюориметрия в ультрафиолетовом свете (предел обнаружения 10-15 г) и эмиссионный спектральный анализ (предел обнаружения 10-12 г).
Некоторые физико-химические методы можно использовать для определений 10-6 – 10-10 г элемента (т. е. 10-6 – 10-10 моль/л). К таким методам относятся, например, спектрофотометрические (атомно-абсорбционная спектрофотомерия и флуориметрия), кинетические и каталитические, а также электрохимические (инверсионная вольтамперометрия) методы анализа.
Методы, основанные на взаимодействии электромагнитного излучения с веществом, бывают обычно более избирательны и имеют более широкую область применения; электрохимические инверсионные методы определения веществ характеризуются более низким пределом обнаружения, кроме того, они требуют более дешевой аппаратуры. В большинстве случаев электрохимические методы менее чувствительны к влиянию основы, чем оптические методы, при гораздо более высокой воспроизводимости и правильности результатов. Электроаналитические методы, в которых используется ртутный электрод, имеют более низкий предел обнаружения при определении некоторых металлов, например при определении Cd, Pb, Tl, Bi. (1)
1.2 Электрохимические методы анализа следовых количеств веществ
Классические электрохимические методы, в основе которых лежит стационарная поляризационная кривая (зависимость стационарного электрохимического тока от напряжения, наложенного на электродную систему), имеют предел обнаружения порядка микрограммов веществ (или ~10-5 моль/л). Такой предел обнаружения определяется отношением электролитического тока (определяемого электродным процессом изучаемого вещества) к остаточному току (сумма электролитических токов примесей, емкостного, обусловленного заряжением электрического двойного слоя тока, и «шума» измерительной цепи). Обычно величина остаточного тока составляет по меньшей мере 10-9 А, поэтому измеряемый электролитический ток должен быть достаточно большим для того, чтобы его можно было отличить от остаточного.
Предел обнаружения классических методов можно снизить, подавляя шум, используя более чувствительную измерительную аппаратуру, измеряя мгновенную концентрацию изучаемого вещества в диффузионном слое у электрода.
Для вращающихся и вибрирующих твердых электродов наряду с проблемами, связанными с достижением воспроизводимой конвективной диффузии, появляются трудности воспроизводимого обновления активной электродной поверхности. В настоящее время наиболее перспективными представляются вращающиеся дисковые электроды, для которых конвективный диффузионный поток электроактивного вещества может быть описан математически и активная поверхность которых с достаточно хорошей воспроизводимостью может быть обновлена полированием.
Чтобы снизить пределы обнаружения электроаналитических методов, необходимо предварительно сконцентрировать разбавленный раствор образца. Для этой цели применимы некоторые методы разделения (хроматография, жидкостная экстракция), при которых происходит отделение мешающих компонентов. Методики продолжительны и трудоемки, возможны потери части определяемого вещества в процессе концентрирования и введение загрязнений в анализируемую систему. Поэтому выгоднее проводить предварительное накопление в системе, в которой проводится измерение. Этот принцип положен в основу электрохимических инверсионных методов: определяемое вещество концентрируется электрохимически на индикаторном электроде (образуя амальгаму или пленку на поверхности электрода), а затем при обратном (электролитическом) процессе переводится в раствор. Таким образом исследуемое вещество в фазе электрода (на границе электрод — раствор) находится в существенно большей концентрации, чем первоначально, и чувствительность определения возрастает во много раз. (1)
1.3 Основы электрохимических инверсионных методов
Электролитическое накопление вещества из разбавленного раствора в большинстве случаев проводится при постоянном потенциале, который выбирается таким образом, чтобы требуемая электродная реакция протекала с достаточной скоростью. Раствор во время электролиза перемешивается, чтобы осуществлялся постоянный перенос деполяризатора из раствора. Для стационарных электродов по истечении определенного времени перемешивание прекращается и раствор успокаивается. За этот период поток вещества к электроду уменьшается, и соответственно величина электролитического тока также быстро падает до величины стационарного диффузионного тока. После стадии успокоения проводится растворение выделенного вещества. При исследовании зависимости тока от электродного потенциала, меняющегося линейно со временем, результирующая поляризационная кривая имеет вид пика, положение которого (потенциал полупика jр/2) характеризует данное вещество, а его высота (или площадь) пропорциональна концентрации вещества в растворе при поддержании постоянных условий предэлектролиза. Схема инверсионной вольтамперометрии приведена на рис. 1.1 и в табл. 1.1.
Таблица 1.1. Основная схема инверсионной вольтамперометрии*
| Стадия | |||
| накопления | успокоения | растворения | |
| Наложенный потенциал | jel = const | jel = const | j = f(t)* |
| Длительность | t | tr | — |
| Ток | Поток деполяризатора | I® IL | IS = f(j) |
*Iel – электролитический ток, IL — предельный ток, IS — ток электролитического растворения.
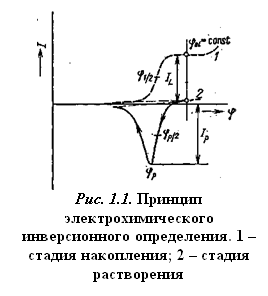
Вольтамперные инверсионные методы называют катодными или анодными в зависимости от характера инверсионного процесса (восстановления или окисления соответственно).
Вообще в инверсионной вольтамперометрии нашли применение две методики работы. Согласно одной из них необходимо полное электролитическое выделение вещества из раствора и контроль тока в течение всего времени, необходимого для полного растворения осажденного вещества. В благоприятных условиях методика позволяет получать правильные и очень хорошо воспроизводимые результаты, хотя длительность определения, особенно при больших объемах раствора, является ее недостатком. При работе с очень малыми объемами образца она все же удобна, так как деполяризатор выделяется из раствора за весьма небольшой промежуток времени (2).
Чаще используется другая методика: накопление проводится в течение определенного времени при воспроизводимых условиях. В этом случае количество осаждаемого на электроде вещества является воспроизводимой долей общего количества вещества в исходном растворе. Методика требует сохранения постоянной скорости переноса вещества к электроду и удобна в тех случаях, если можно подобрать условия предварительного электролиза, чтобы осаждаемая доля составляла только 2 – 3 % от общего количества вещества.
Высота пика растворения обычно зависит от следующих факторов:
а) количества вещества, осажденного на электроде, которое является функцией его концентрации в растворе, потенциала накопления, продолжительности накопления, скорости потока вещества из объема раствора к электроду (т. е. интенсивности перемешивания или скорости вращения электрода), площади активной поверхности электрода, состава раствора, температуры и электрохимических свойств системы;
б) условий процесса растворения, особенно от скорости поляризации, площади активной поверхности электрода, скорости отвода продуктов от электрода.
Если суммарный электродный процесс включает и химическую реакцию, то на высоту пика оказывают влияние скорость этой реакции, характер продуктов реакции, растворимость образующихся соединений и т. п.
1.4 Реакции, используемые для электролитического накопления
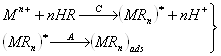
Для накопления вещества могут быть использованы различные электрохимические и химические реакции, например: восстановление катионов до соответствующего металла, образование амальгамы или малорастворимого соединения, адсорбция. Для определения веществ различных классов существуют определенные типы реакций и в соответствии с природой образующегося осадка подбираются остальные условия, и, прежде всего рабочий электрод. Чаще всего используются перечисленные ниже типы реакций.
2. Металлы, способные образовывать достаточно концентрированные амальгамы, могут быть сконцентрированы на стационарном ртутном электроде. Металл, образовавшийся из ионов при их электровосстановлении, растворяется в ртутном электроде; затем он анодно растворяется из амальгамы; регистрируется анодный ток.
3. Ионы металла могут быть восстановлены до металла и накоплены на подходящем инертном электроде (благородный металл, графит) в виде пленки. Этот процесс наиболее часто используется для определения ртути, благородных металлов и металлов, не образующих амальгам. Определяемое вещество можно сконцентрировать на электроде в виде малорастворимого соединения. По способу образования последнего реакции подразделяют еще на два типа:
а) Малорастворимое соединение образуется при взаимодействии с ионами материала электрода. Такое соединение концентрируется на электроде при потенциале, соответствующем окислению электродного материала, затем регистрируется катодное растворение пленки (например, определение хлорид-иона на ртутном, или серебряном электроде). Подобные реакции можно использовать для косвенного определения некоторых металлов. Косвенное определение серебра или золота с применением стационарного электрода можно проводить в растворе, содержащем сульфид-ион. После добавления ионов серебра или золота регистрируемый первоначальный пик, соответствующий катодному растворению сульфида, уменьшается, так как часть сульфид-иона осаждается в виде малорастворимого сульфида серебра или золота.
б) Малорастворимое соединение образуется в виде пленки на электроде при взаимодействии с некоторыми компонентами основного электролита или с реагентом, добавляемым в раствор. Во время электродной реакции ионы определяемого вещества восстанавливаются или окисляются до степени окисления, в которой они участвуют в химической реакции, приводящей к образованию осадка. Таким образом, в химической реакции с реагентом принимают участие только ионы, полученные в результате электродной реакции; кроме того, образование осадка должно происходить быстрее, чем перенос продукта электродной реакции от электрода в объем раствора. Только при этих условиях количество образовавшегося осадка пропорционально концентрации вещества в растворе. Самым простым примером применения этой реакции является определение некоторых элементов, образующих малорастворимые оксиды, например определение марганца или свинца после окисления ионов Мn2+ и Рb2+ до МnО2 и РbО2.
4. Для предварительного концентрирования некоторых ионов можно использовать поверхностно-активные вещества. Эти вещества могут адсорбироваться на поверхности электрода и реагировать с определяемым ионом с образованием комплекса (тип 4а), или комплекс иона с поверхностным веществом образуется в растворе и затем адсорбируется на поверхности электрода (тип 4б). Электродная реакция при растворении сводится к восстановлению или окислению адсорбированного на электроде комплекса. Важно, что при этом типе реакций можно достичь накопления даже в отсутствие электролитического тока, хотя количество адсорбированного вещества часто зависит от наложенного на электрод потенциала.
Кроме этих четырех основных типов описаны различные косвенные определения, основанные, например, на вытеснении электроактивного металла из его комплекса электронеактивным металлом или на реакциях осаждения (примеры приведены при рассмотрении третьего способа).
Таблица 1.2. Реакции, используемые в методе инверсионной вольтамперометрии
| Тип реакции | Вещество в растворе | ПРЕДВАРИТЕЛЬНОЕ НАКОПЛЕНИЕ | РАСТВОРЕНИЕ |
| 1 | Mn+ |
|
|
| 2 | Mn+ |
|
|
| 3а | An- |
|
|
| 3б | Mn+ |
|
|
| 4а | Mn+ |
|
|
| 4б | Mn+ |
|
|
| 5 | Mn+ |
|
|
| · | An- |
|
|
| 6 | Mn+ |
|
|
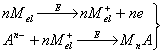

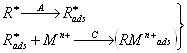
* ![]() - материал электрода; А – адсорбция; * - поверхностно-активные вещества; Е – электродная реакция; Ех – экстракция; С – химическая реакция; I – ионный обмен; индексы: w - водный; org – органический; ads – адсорбированный; ion – сорбированный на ионообменнике.
- материал электрода; А – адсорбция; * - поверхностно-активные вещества; Е – электродная реакция; Ех – экстракция; С – химическая реакция; I – ионный обмен; индексы: w - водный; org – органический; ads – адсорбированный; ion – сорбированный на ионообменнике.
Экстракция адсорбированного на поверхности электрода вещества в малый объем растворителя.
Концентрирование на ионообменной мембране, размещенной непосредственно на поверхности электрода.
В табл. 1.2 кратко приведены рассмотренные выше реакции. Уравнения представляют собой только схемы процессов.
Предел обнаружения инверсионных методов можно улучшить при использовании каталитических реакций. В этом случае соединение, электродная реакция которого катализируется накопленным веществом, находится в растворе, и его каталитический ток измеряется в стадии растворения (3).
Для понижения предела обнаружения, улучшения правильности и воспроизводимости большое значение имеет выбор рабочего электрода. Используемые электроды можно разделить на две группы: ртутные и твердые.
Самым простым, наиболее часто употребляемым типом ртутного электрода является стационарный ртутный капельный электрод. Такой электрод состоит из капилляра, соединенного с резервуаром, ртутная капля выдавливается из капилляра и стабилизируется у его устья. В самых распространенных устройствах капилляр вертикален и капля «подвешена» на ртутном столбике. Капилляр можно также изогнуть устьем вверх («лежащая» капля); такая конструкция дает определенные преимущества, так как улучшается стабильность капли и появляется возможность использовать капилляры с меньшим внутренним диаметром, что предотвращает обратную диффузию металла в капилляр. Кроме нестабильности капли в обратной диффузии, существенным недостатком таких стационарных ртутных электродов является проникновение раствора внутрь капилляра.
Как правило, в стационарном ртутном капельном электроде ртутная капля либо механически «подвешивается», либо осаждается электролитически на небольшом контакте из инертного металла (серебра, платины или золота). Вся поверхность контакта должна быть покрыта ртутью, а материал контакта должен иметь минимальную растворимость в ртути. При конкретных определениях все же необходимо учитывать возможность образования интерметаллических соединений определяемого вещества, с материалом контакта, особенно при использовании контакта из золота.
В последнее время все чаще применяется ртутный пленочный электрод: пленка ртути электролитически осаждается на благородный металл либо на графит или углерод. При использовании таких электродов можно снизить предел обнаружения, ибо у таких электродов отношение активной поверхности к объему ртути значительно возрастает (толщина пленки часто соответствует лишь нескольким молекулярным слоям). Кроме того, увеличивается разрешающая способность, так как диффузия металла в фазе электрода весьма ограничена и пики растворения получаются значительно более узкими. Пленочные ртутные электроды могут быть вращающимися, например вращающийся дисковый электрод.
Воспроизводимость результатов при работе со стационарными ртутными электродами зависит от воспроизводимости размера капли (или воспроизводимости массы ртути на твердой подложке). На практике не сложно достигнуть воспроизводимости, поэтому работать с ртутными электродами обычно очень удобно. Если образуется жидкая амальгама, то можно предположить, что осажденное вещество имеет одинаковую активность во всем объеме электрода. При анализе большинства смесей металлы, находящиеся в сравнимых концентрациях, не оказывают взаимного влияния друг на друга. Высокое перенапряжение выделения водорода позволяет определять металлы, дающие волны восстановления при весьма отрицательных потенциалах. В нейтральных растворах рабочая область потенциалов находится в интервале от —2,5 до +0,2 В.
Твердые электроды из платины, серебра, золота или графита используются при работе в области положительных потенциалов, где наблюдается растворение ртути.
При изготовлении электродов обычные графитовые материалы необходимо импрегнировать соответствующим способом (чтобы не происходило проникновения раствора в поры) или использовать угольные пасты. Пиролитический графит и стеклоуглерод не требуют импрегнирования.
При электролизе с твердыми электродами пленка образуется на поверхности электрода; это приводит к возникновению более сложной ситуации, чем в случае амальгамных электродов. Образование и растворение поверхностных пленок подчиняется более сложным зависимостям; на них оказывают влияние, например, структура и поверхностная энергия электрода, поверхностные каталитические явления, структура образовавшегося осадка. При анализе смесей часто наблюдаются взаимные помехи, и практически достигаемые пределы обнаружения часто гораздо выше, чем с применением ртутных электродов. Для получения воспроизводимых результатов необходимо, чтобы площадь активной поверхности электрода была постоянна и воспроизводимо обновлялась. Эта проблема решается специфично для каждого случая. Поверхностный слой оксидов на металлических электродах, например платиновом, может оказывать значительное влияние на электродную реакцию. Этот эффект следует изучить прежде, чем приступить к собственно определению. Несмотря на отмеченные недостатки, твердые электроды существенно расширяют возможности инверсионных методов и в настоящее время интенсивно изучаются.
Угольный пастовый электрод является особым типом твердого электрода. При соответствующей конструкции электрода можно выдавливать угольную пасту подобно тому, как выдавливается ртутная капля у стационарных ртутных электродов. Этим способом можно относительно легко обновлять активную поверхность, добиваясь достаточной воспроизводимости. Для приготовления угольной пасты используется подходящий органический растворитель или силиконовое масло. В некоторых случаях эти вещества могут, принимать участие в реакциях, которые происходят при предварительном концентрировании определяемых соединений. Например, при определении бромидов или броматов можно электролитически получить бром, который легко растворяется (и концентрируется) в растворителе, входящем в угольную пасту, но затем трудно добиться его количественного растворения из электрода. (1)
1.6 Методы исследования процесса растворения
Принцип накопления вещества и последующего его электрохимического растворения не является новым; он был, например, использован для измерения толщины металлических пленок. Збинден (4) уже в 1931 г. определял следовые количества меди, осаждая ее на платиновом электроде и измеряя зависимость анодного тока от времени при соответствующем постоянном потенциале в процессе растворения пленки металла.
В пятидесятых годах прием электролитического накопления и последующего электрохимического растворения вещества был распространен на многие электрохимические методы. Наибольшую известность получила вольтамперометрия с линейным изменением потенциала во времени (5 – 7) в связи с ее методической и инструментальной простотой.
Осциллографическая полярография (8) (рабочий электрод поляризуется переменным током с постоянной плотностью, амплитудой и частотой, а на экране осциллографа регистрируется функция dj/dt = f(j), квадратноволновая полярография (9) и переменнотоковая полярография (10), хронопотенциометрия и кулонометрия (11 – 13) могут быть также использованы для исследования процесса растворения. В некоторых случаях для повышения чувствительности определения применяют нестационарные методы. Для исследования процесса электрохимического растворения используются, таким образом, любые методы, основанные на изучении стационарных и нестационарных поляризационных кривых (табл. 1.3).
Таблица 1.3. Методы, применяемые при исследовании инверсионного процесса 3
| Контролируемый параметр | Измеряемая функция | Название метода |
| Стационарные методы | ||
| j | I = f(j) | Вольтамперометрия при постоянном потенциале |
| j |
| Кулонометрия при постоянном потенциале |
| j | Q = f(c) | Полярографическая кулонометрия |
| I | Q = It | Кулонометрия при постоянном токе |
| Нестационарные (потенциостатические) методы | ||
| j | I = f(t) | Хроноамперометрия |
| j = ji + wt | I = f(j) | Полярография и вольтамперометрия с переменным потенциалом (single sweep, multi-sweep) |