


Стандартизация измерения рН в неводных средах. Методы определения рН стандартных буферных растворов
Сложность явлений, происходящих в растворах, а также необходимость изучения растворов с широким диапазоном физических и химических свойств растворителя неизбежно привлекали внимание исследователей к неводным средам. Неводные растворители постепенно начали использовать не только для научных исследований, но и в практической деятельности. Они широко применяются в аналитической практике для улучшения условий титрования. Неводные растворители часто используются в химической, фармацевтической, нефтеперерабатывающей промышленности, в промышленности высокополимеров и в других отраслях народного хозяйства.
Быстрыми темпами осуществляется переход к проведению процессов органического синтеза в неводных средах. Замена воды органическими растворителями и переход на замкнутые системы производства позволяет, во-первых, резко интенсифицировать химические процессы, во-вторых, упрощает решение проблем, связанных с очисткой сточных вод и с нарастающей нехваткой пресной воды. Поэтому понятен интерес к методам оценки кислотности неводных растворов и растворов в смешанных растворителях.
Используемые в качестве эталонов для измерений буферные растворы введены давно. Стандартные буферные растворы используются для калибровки приборов, поэтому точность определения кислотности этих растворов должна быть высокой.
1.Понятие pH
Водородный показатель, pH — это мера активности(в случае разбавленных растворов совпадает с концентрацией) ионов водорода в растворе, количественно выражающая его кислотность, вычисляется как отрицательный десятичный логарифм концентрации водородных ионов, выраженной в молях на литр:
![]() (1)
(1)
Зависимость громадного количества химических, биохимических, природных, технологических и многих других процессов от величины рН стимулирует развитие теории и техники измерения этого показателя.
Трудно отыскать такую область науки, технологии и химии растворов, в которой не использовалась бы величина рН. Шкала рН была предложена датским химиком Зеренсеном. Изучая биохимические реакции, в частности гидролиз пепсина и усвоение протеинов в кислотно-солевых смесях, Зеренсен в 1909 году обнаружил их чрезвычайно сильную зависимость от изменения концентрации ионов водорода. Он нашел, что эффективная концентрация ионов водорода, непосредственно воздействующих на процесс, изменялась в широком диапазоне концентраций и часто оказывалась непривычно малой величиной. Для удобства Зеренсен предложил записывать ее в экспоненциальной форме сН+ = 10-р = 1/10р. Позднее символ -р был заменен обозначением рН. На современном языке р понимают как оператор р = -lg, таким образом, pH = -lg(H+). Именно так трактуется это обозначение в Номенклатурных правилах Международного союза по теоретической и прикладной химии (International Union for Pure and Applied Chemistry), ИЮПАК.
2.Кислотность неводных растворов.
2.1.Шкала рНр
Важным вопросом является определение рН в неводных и смешанных растворителях. Этот вопрос имеет практическое значение, так как в пищевой промышленности, промышленности пластмасс, фото- кино- промышленности и других отраслях промышленности широко используют измерения рН в неводных растворах.
При определении рН в неводных растворах делается еще большее количество ошибок, чем при определении рН в водных растворах.
При решении проблемы о кислотностях неводных растворов следует поставить два вопроса. Как поступать при сравнении кислотности двух растворов в одном и том же растворителе? Как поступать присравнении кислотности растворов в двух разных растворителях? Эта задача отличается принципиально от задачи сравнения между собой кислотности в пределах одного растворителя.
Очень часто намерении pН в неводных растворах производят по отношению к водному каломельному электроду, потенциал которого определяется по водному стандарту. При этом фактически измерения сводят к измерению э.д.с цепи:
Pt(H2) | стандартный | исследуемый | Pt(H2) (2.1.1)
р-р в воде р-р в неводн р-ле
Таким образомзначения рН неводных растворов не могут быть получены путем непосредственного сопоставления потенциалов индикаторных электродов в водных и неводных растворах, а следовательно, и рН-метр, калиброванный по водным стандартам, не может дать правильных значений рН неводного раствора.
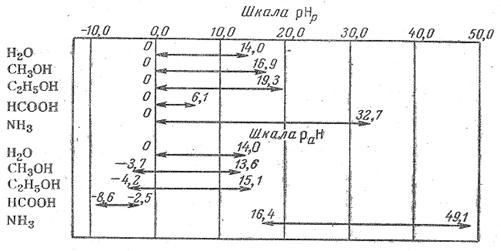
Рис. 1. Шкала рНр и шкала раН в различных растворителях.
По вопросу о стандартизации рН в неводных растворах нет единого мнения. Тем не менее расширение этого вопроса становится все более насущным в связи с широким распространением неводных растворителей, и особенно их смесей с водой, в промышленности и аналитической практике.
Задача сравнения кислотности в пределах одного неводного растворителя принципиально не отличается от задачи определения рН в водных растворах. Величина рН определяется отрицательным логарифмом активности ионов водорода в данном растворителе М:
pHp=-lga*=-lgc![]() *
*
т.е. отрицательным логарифмом произведения концентрации ионов лиония
на соответствующий концентрационный коэффициент активности. Коэффициент активности относится к бесконечно разбавленному раствору в данной среде как стандарту.
Переходя от воды в неводному растворителю, переходят от кислотности, выраженной в концентрациях или активностях одного иона - иона гидроксония, к кислотности, выраженном в концентрациях или активностях другого иона – лиония. Действительно, в воде носителем кислотных свойств является ион Н3О+. Когда по отношению к водному раствору мы говорим, что его рН = 5, это значит, что активность ионов Н3О+ равна 10-5.В спиртовом растворе носителем кислотных свойств являются ионы этоксония С2Н5ОН2+, в аммиаке носителем кислотных свойств — соответственно ионы аммония NH4+ и т. д.
При оценке рН растворов в пределах одного растворителя следует учитывать протяженность шкалы рН: в.воде вся шкала рН равна 14,0, но в этиловом спирте 19,3; в муравьиной кислоте. 6,1 и т. д. (рис. 1)
Стандартизация рН в неводных растворах может быть выполнена так же как и в водных растворах, т. е. путем изготовления стандартных растворов в том же растворителе, что и исследуемый раствор. Однако в этом случае возникает ряд затруднений. Например, коэффициенты активности сильных кислот значительно больше отличаются от единицы, чем в водных растворах; сильные в воде кислоты становятся в неводных растворах слабыми; хуже растворимы соли; значительно меньше имеется данных о коэффициентах активности.
В настоящее время единственным веществом, с помощью которого может быть произведена стандартизация рН в неводных растворах, является хлористый водород, так как для него имеются данные о коэффициентах активности в большинстве широко используемых растворителей и в их смесях с водой. В качестве электрода сравнения при измерениях в неводных растворах может быть использован хлорсеребрянный электрод в растворе НCl, который вполне пригоден для измерений в ряде чистых неводных растворителей и их смесях с водой.
Измеренная по отношению к стандарту в данном растворителю рН не является абсолютном мерой кислотности неводного раствора и может быть использована для характеристики кислотности только в пределах данного растворителя. Это следует из того, что начало шкалы кислотности раНр =0 не соответствует равенству абсолютных активностей ионов водорода во всех растворителях. Величины раН нейтральных растворов в разных растворителях не совпадают друг с другом, так как протяженность шкал, зависящая от ионного произведения растворителя, различна. В верхней части рис. 1 в качестве примера приведены шкалы рНр в воде и не которых неводных средах. В воде шкала рН изменяется от 0 до 14; нейтральным раствором называется раствор с рН = 7. Если раствор имеет рН=0, это раствор кислоты с активностью ионов Н+, равной единице; если раствор имеет рН = 14, это раствор щелочи с активностью ионов ОН--, равной единице, но это не значит, что не может быть растворов в воде с рН меньше нуля и больше 14.
Раствор, у которого рН =15, означает раствор щелочи с активностью, равной 10, растворы с рН=-1 являются растворами кислоты с активностью, равной 10.
В воде между одноактивным раствором кислоты и одноактивным раствором щелочей изменение рН равно 14, это является следствием того, что ионное произведение воды равно 10-14. Вметиловом спирте ионное произведение равно 10-16,5. Это значит что между одноактивным раствором щелочи и одноактивным раствором кислоты изменение рН будет составлять 16,5 единицы. У нейтрального раствора рН будет не 7, как в воде, а 8,3. Этиловый спирт имеет шкалу протяженностью 19,3 единицы; следовательно, рН нейтрального раствора будет составлять 9,7. В аммиаке шкала имеет протяженность 32,7 единицы и нейтральный раствор в нем будет иметь рН = 16,3.
Таким образом, нужно иметь в виду, как относится величина рН данного раствора к рН нейтрального раствора.
Поэтому предложено отмечать показатель рН индексом, равным рН нейтральной точки. В воде рН-следует отмечать индексом 7 - рН7, в этиловом спирте индексом 9,7 — рН9,7 и т. д.
Мы уже говорили о невозможности определения рН в цепях, содержащих водный и неводный растворы. Измерение рН должно производиться в цепи, содержащей только один растворитель; стандартный и измеряемый растворы должны быть в одном и том же растворителе. При измерении рН в спирте стандартные растворы должны быть также спиртовыми.
Это положение не исключает практического применения водного стандарта. Можно пользоваться водным каломельным электродом и в этих случаях, но его потенциал следует измерять по отношению к неводному стандарту. В принципе стандартизация шкалы рНр всегда должна быть произведена по отношению к неводному раствору ионов водорода.
2.2 Единая шкала кислотности
Для решения ряда практически важных вопросов возникает необходимость сопоставления кислотности растворов в различных растворителях, приведение значений рНр к единому началу отсчета. Можно было бы полагать, что измерения, произведенные на рН-метре, откалиброванном по водным стандартам, должны давать значения рН по отношению к единому стандарту, так как электрод сравнения остается неизменным и измеряемая э.д.с представляет собой разность потенциалов электродов, обратимых но отношению к ионам водорода, опущенных в стандартный водный иисследуемый неводный растворы. Однако, как уже говорилось, наличие фазового потенциала не позволяет находить значения раНр, отнесенные к единому водному стандарту.
Как же сравнивать кислотность в двух различных растворителях? Как решить вопрос о том, какой раствор кислее — водный с рН = 3 или спиртовой с тем же рНр — 3? Вопрос о сопоставлении кислотности представляет большие трудности как принципиального, так и экспериментального характера. Эти затруднения пытались решать разными методами. Самой правильной является постановка вопроса о кислотности неводных растворен Бренстеда. Бренстед предлагает во всех растворах считать мерой кислотности абсолютную активность протона или величину, ей пропорциональную — химический потенциал протона:
![]() (2.2.1)
(2.2.1)
Принципиально определение абсолютной активности протона вполне возможно. Константа собственной кислотности кислоты равна активности протона, умноженной на активность основания и деленной на активность кислоты. При этом речь идет об абсолютной активности, т. е. об активности, отнесенной к единому стандартному состоянию:
Ka=aH+*aB/aA(2.2.2)
Заменяя абсолютные активности произведениями активностей, отнесенных к бесконечно разбавленному раствору в данной среде М, и единых коэффициентов активности ץ 0 получим выражение
Ka=![]() (2.2.3)
(2.2.3)
Откуда: aH+=![]() (2.2.4)
(2.2.4)
Из этого выражения вытекает, что если бы можно было действительно найти константу собственной кислотности какой-то кислоты в вакууме и![]() и
и ![]() , то можно было бы определить активность протона. Величины
, то можно было бы определить активность протона. Величины ![]() и
и ![]() можно определить экспериментально. Можно найти, сколько в данном растворителе кислоты и сколько основания, т. е. установить, в какой степени данная кислота продиссоциирована, найти концентрационные коэффициенты активности
можно определить экспериментально. Можно найти, сколько в данном растворителе кислоты и сколько основания, т. е. установить, в какой степени данная кислота продиссоциирована, найти концентрационные коэффициенты активности ![]() и
и ![]() и найти активность а*.
и найти активность а*.
Основные затруднения состоят в определении единых коэффициентов активности ![]() кислоты и соответственно основания
кислоты и соответственно основания ![]() .
.
Мерой этих абсолютных коэффициентов активности является энергия переноса вещества из вакуума в данную среду, т. е. энергия их сольватации.
Следовательно, для того чтобы использовать в качестве единой меры кислотности активность протона, нужно знать собственную константу кислотности кислоты и знать энергии переноса кислоты и основания из вакуума вданную среду.
В настоящее время эти величины известны только очень приближенно, поэтому такой путь определения истинной активности протона еще не может быть осуществлен.
Если известно протонное сродство (работа присоединения протона к данному веществу в вакууме), то из него всегда можно вычислить константу собственной кислотности. Следовательно, некоторые возможности определения активности протона этим путем уже намечаются. Если раньше константа собственной кислотности была фиктивной мерой силы кислоты, то сейчас ее можно рассматривать как реальную меру. Применение этого метода затруднено только недостаточной точностью в определении протонного сродства и ![]() .
.
2.3 Метод Михаэлиса. Шкала рНHAc Конанта и Хелла
Трудности в определении активности протона привели к тому, что было предложено много других методов оценки кислотности в неводных растворах. Первой была попытка Михаэлиса и Митцутани, которые предложили оценивать кислотность в неводных растворах, измеряя э. д. с. цепи, включающей диффузионный и фазовый потенциал
Pt(H2) | H+ в воде | | Н+в М | Pt(H2) (2.3.1)
и на основании э. д. с. этой цепи вычислять кислотность. Если бы не было дифузионного, и особенно фазового, потенциалов, то эту цепь, действительно, можно было бы применять для оценки кислотности, потому что потенциалы электродов различаются настолько, насколько различаются между собой активности ионов лиония в этих двух растворах. На самом доле э. д. с. этой цепи определяется не только разностью активности ионов лиония, но и величиной фазового потенциала, который обычно направлен против потенциала, обусловленного различной активностью протонов. Поэтому метод Михаэлиса и Митцутани непригоден для оценки абсолютной кислотности.
Конант и Хелл, исследуя кислотность в уксусной кислоте как растворителе, предложили измерять кислотность в такой цепи:
Ptхлоранил| Н+ в уксусной кислоте|| KCl в воде| Hg2Cl2,Hg (2.3.2)
Хлоранил представляет собой эквимолекулярную смесь С6(ОН)2С14 и С6С1402. С помощью этого вещества можно измерять кислотность очень кислых растворов. В воде потенциал хлоранилового электрода против каломельного равен 0,418 В. Конант и Хелл для своей цели приняли, что потенциал хлоранилового электрода против каломельного равен не 0,418, а 0,566. Они считали, что разница на 0,148 В соответствует фазовому потенциалу,который возникает на границе уксусной кислоты и водного раствора, и изменению нормального потенциала хлоранилового электрода. Но это предположение произвольно. Эта разница очень плохо оправдана. Конант и Хелл приняли ее на том основании, что в результате введения поправки константа диссоциации пиридина в уксусной кислоте равна константе диссоциации у уксусной кислоты в воде. Равенство констант принято ими на основании изучения электропроводности растворов. Однако это предположение сомнительно.
Кислотность, определенную по Конанту и Хеллу, принято обозначать pHHAc.
2.4 Определение кислотности методом Гамметта
Основываясь на том, что, как свидетельствуют экспериментальные данные, константы кислотности оснований (катионных кислот) сравнительно-мало изменяются при переходе от растворителя к растворителю, Гамметт предложил оценивать кислотность любых растворов по степени превращения индикатора основания в его ионную форму.
Известно, что величина рН водных растворов может быть определена про помощи индикаторов. В основе индикаторного метода лежит уравнение
pH=pK+lg(aAi/aHAi) (2.4.1)
где aAi и aHAi активности ионной и молекулярной формы индикатора.
В случае, если индикатором является основание, уравнение приобретает вид:
pH=pK+lg(aBi/aBHi) (2.4.2)
Различия в окраске основания и катионной кислоты, соответствующей этому основанию, или кислоты и аниона этой кислоты позволяют установить кислотность. Метод основан на том, что по окраске оценивают концентрацию кислой и основной форм индикатора. Сравнение окраски в данном растворе с окраской раствора, содержащего предельную форму индикатора в условиях, когда индикатор полностью превращен либо в кислоту, либо в основание, производится в колориметре. Особенно удобны для этих целей одноцветные индикаторы, у которых одна из форм окрашена, а другая не окрашена.
Не будем подробно останавливаться на методике индикаторного определения рН. Отметим только, что при правильном осуществлении этот метод определения рН достаточно точен. Однако применение индикаторного метода не исключает ошибок, связанных со стандартизацией рН. Кроме того, индикаторный метод имеет ряд специфических ограничении, с которыми следует считаться.
Во-первых, если раствор содержит окислители или восстановители, то пользоваться колориметрическим методом следует с осторожностью, так как при этом может произойти окисление индикатора, и окраска (и ее интенсивность) будет изменяться не за счет изменения рН, а за счет окисления индикатора. К тому же многие вещества одновременно являются кислотно-основными и окислительно-восстановительными индикаторами и реагируют на наличие в растворах окислительно-восстановительных систем.
Во-вторых, индикаторы ограниченно применимы в небуферных системач, так как каждый индикатор — это или кислота., или основание, и прибавление их к небуферным системам создает определенную кислотность. В этих случаях фактически измеряется та величина рН, которая создалась в результате растворения индикатора.
В-третьих, окраска индикатора изменяется в зависимости от ионной силы раствора.
В-четвертых, многие индикаторы реагируют с белками, поэтому в белковых системах, в биологических средах индикаторный метод может при вести к так называемым белковым ошибкам.
Возвратимся к основному вопросу — к определению единой кислот ности. Согласно Гамметту, окраска одного индикатора изменяется в различных растворителях только в связи с изменением абсолютной кислотности растворов, а константа индикатора основания в любом растворителе остается неизменной. Соотношение основной и кислой форм индикатора изменяется только в связи с изменением кислотности раствора. Свою функцию кислотности Гамметт обозначает Н0, так как индикаторы основания не имеют электрического заряда. По Гамметту
Н0=pKa+lg(cB/cBH+) (2.4.3)
где pKa - показатель константы диссоциации индикатора как катионной кислоты в воде. Эта константа принимается неизменной.
В дальнейшем были введены другие функции кислотности. В тех случаях, когда применяется в качестве индикатора незаряженная кислота и соответствующее ей основание имеет отрицательный заряд, функцию кислотности обозначают Н(-).
Метод Гамметта чрезвычайно прост и не связан с измерением потенциалов, не имеет осложнений в связи с возникновением потенциалов на границе двух фаз. Поэтому он представляет значительный интерес и нашел широкое применение.
Однако последние работы показали, что нет оснований считать, что в действительности величина Н0 передает кислотность неводных растворов. Предположение о том, что константа индикатора не изменяется при переходе от растворителя к растворителю, очень сомнительно.
Предположение Гамметта о неизменности констант кислотности индикаторов-оснований равносильно предположению, что константы кислотности оснований выражены через абсолютные активности, отнесенные к водному раствору как к стандарту.
Искомой величиной является абсолютная активность ионов лиония аМН+ отнесенная к водному раствору протонов (ионов гидроксония) как к стандарту. Константа кислотности основания через абсолютные активности выразится так:
КАосн=аВ+(М)(aB/aBH+) (2.4.4)
Заменив в уравнении (2.4.4) величины aBи aBH+ выражениями a=c![]() получим:
получим:
KAосн=аH+(M)(cB![]() (2.4.5)
(2.4.5)
где аH+(M) искомая абсолютная активность сольватированного протона, отнесенная к его состоянию в бесконечно разбавленном, водном растворе.
На основании уравнения (2.4.5) для КАосн запишем выражение для рКА:
pKA=-lgKAосн=-lgaH+(M)(cB![]() (2.4.6)
(2.4.6)
Подставив это выражение в уравнение (2.4.3), получим:
![]() (2.4.7)
(2.4.7)
Предположение Гамметта будет действительно правильным, если окажется, что Н0будет равно только логарифму активности протонов в данном растворителе, отнесенному к единому стандартному состоянию, т. е. к бесконечно разбавленному водному раствору. Но для этого нужно, чтобы выражение ![]() было равно нулю.
было равно нулю.
Упростим нашу задачу: представим, что растворы настолько разбавлены, что отношение концентрационных коэффициентов активности ![]() единице. Но и тогда в выражении остается отношение единых коэффициентов активности
единице. Но и тогда в выражении остается отношение единых коэффициентов активности ![]() они не зависят от концентрации. В этом случае выражение для Н0 примет вид:
они не зависят от концентрации. В этом случае выражение для Н0 примет вид:
Н0=![]() (2.4.8)
(2.4.8)
Из этого выражения следует, что Н0не равно логарифму активности протона, а отличается от него на величину логарифма отношения коэффициентов активности ![]() заряженной и незаряженной форм индикатора т. е. зависит от того, какова энергия взаимодействия с растворителем иона и нейтральной молекулы индикатора. При стандартизации по отношению к бесконечно разбавленному водному раствору величины
заряженной и незаряженной форм индикатора т. е. зависит от того, какова энергия взаимодействия с растворителем иона и нейтральной молекулы индикатора. При стандартизации по отношению к бесконечно разбавленному водному раствору величины ![]() и
и ![]() определяются работой переноса ионов ВН+ и соответственно молекул Виз среды М в воду. Таким образом, предположение, что Н0равно -lgaH+(M) будит справедливо только в том случае, если влияние растворителя на катион основания и молекулу основания индикатора одинаково.
определяются работой переноса ионов ВН+ и соответственно молекул Виз среды М в воду. Таким образом, предположение, что Н0равно -lgaH+(M) будит справедливо только в том случае, если влияние растворителя на катион основания и молекулу основания индикатора одинаково.
Но все количественные данные, имеющиеся по этому поводу, говорят о том, что это не так.
Шварценбах пытался сравнить ход изменения величины Н0и величины Н(-)в зависимости от свойств растворителей. В соответствии со сказанным ранее величина Н(-)будет выражена через единые коэффициенты активности так: ![]() (2.4.9)
(2.4.9)
По Шварценбаху, аналогия заключается в том, что коэффициенты активности ![]() и
и ![]() ,так же как и коэффициенты активности
,так же как и коэффициенты активности ![]() и
и ![]() представляют коэффициенты активности веществ, отличающихся только одним зарядом.
представляют коэффициенты активности веществ, отличающихся только одним зарядом.
Предположение Шварценбаха сводится к тому, что отношение коэффициентов ![]() /
/ ![]() равно отношению
равно отношению ![]() /
/![]()
Оказалось, что это далеко не так. Исследование этих функции кислотности в 0,002 н. раствора НС1 в смесях спирта с водой показало для функции Н0иную зависимость от содержания спирта, чем для функции Н(-). Следовательно, Н(-) не передает истинной кислотности раствора. Появление заряда на молекуле осложнит в результате присоединения протона вызывает также резкое изменение энергии взаимодействия незаряженной молекулы индикатора и ее иона с растворителем. Все это говорит о том, что нельзя приравнивать изменение H0к изменению кислотности. Задача может быть решена, если будут известны ![]() для В и ВН+, только тогда Н0можно исправить и найти истинную величину -lg аН+(М).
для В и ВН+, только тогда Н0можно исправить и найти истинную величину -lg аН+(М).
Таким образом, ни Н0, ни Н(-)не оценивают правильно единую кислотность растворов.
Очень существенным недостатком метода Гамметта является также необходимость использования нескольких индикаторов при определении кислотности. Согласно теории индикаторов, заметить изменение окраски, по которой судят об изменении Н0, можно только тогда, когда будет присутствовать не меньше 10% одной формы индикатора в присутствии 90% другой; можно заметить окраску вещества ВН+, когда оно будет составлять более 10% от вещества В, и наоборот. С помощью одного индикатора можно определить изменение кислотности только в пределах 2 единиц рН и Н0.
В воде шкала рН=14; следовательно, нужно иметь семь индикатором для оценки кислотности.
Рассмотрим, насколько метод Гаммета пригоден для определения кислотности в пределах одного неводного растворителя.
Нельзя принимать, как это делает Гамметт, что в неводных растворах соотношение между константами индикаторов остается таким же, как и в воде. Как известно, растворитель оказывает дифференцирующее действие. Оно сводится к тому, что относительная сила кислот или оснований изменяется при переходе от одного растворителя к другому.
Разность в pК двух индикаторов основания В1и В2 определится выражением:
pKA1-pKA2=-lg![]()
![]()
![]()
![]() (2.4.10)
(2.4.10)
Введем концентрационные активности а* и коэффициенты активности ![]() тогда получим:
тогда получим:
pKA1-pKA2=-lg![]() (2.4.11)
(2.4.11)
Из уравнения (26) следует, что разность рK определяется не только соотношением активностей а*, но и соотношением коэффициентов активности ![]() . Нет никаких оснований утверждать, что
. Нет никаких оснований утверждать, что
![]() (2.4.12)
(2.4.12)
и, следовательно, соотношение рКА не остается неизменным.
Это обстоятельство затрудняет использование метода Гамметта и в пределах одного растворителя. Необходима экспериментальная проверка pKиндикаторов в каждом растворителе.
Есть и третий недостаток метода Гамметта, заключающийся в том, что иногда окраска индикатора изменяется не в связи с изменением соотношения между разными формами индикаторов ВН+ и В, а в связи с тем, что окраска одной из форм индикатора изменяется под влиянием растворителя. Однако главный недостаток метода Гамметта состоит в том, что влияние растворителей па заряженную и незаряженную формы индикатора не одинаково, в связи с чем Н0 не передает истинной кислотности неводных растворов.
Для оценки кислотности кроме функций Н0 и Н(-) предложены функция Н(+), основанная на зависимости положения равновесия реакции ВН2+= В+ + Н+ от кислотности, а также функция кислотности I0 , основанная на зависимости положения равновесия реакции ROH + H+ =R+ + H2O (R+ -ион карбония, ROH-арилкарбинол) от кислотности.
Каждая функция кислотности определяется значением соответствующих величин рК и отношением концентраций кислотной и основной форм индикатора:
H0=pKBH++lg(cB/cBH+) H(-)=pKBH+lg(cB-/cBH)
I0=pKR++lg(cROH/cR+) H(+)=pKBH2++lg(cB+/cBH2+)
Соотношение между этими функциями кислотности и величиной истинной единой кислотности рА = -lg aH+ определяется следующими выражениями:
![]()
![]()
![]()
![]()
из которых следует, что они не совпадают между собой и что ни один из них не передает истинной кислотности.
2.5Метод нормального потенциала Плескова
Исследуя потенциалы щелочных металлов — лития, натрия, калия , рубидия, цезия, - Плесков установил, что э. д. с. цепи Rb|Rb+||Cs+|Cs оказывается неизменной во многих растворителях. На основании этого Плесков высказал предположение о том, что потенциал цезиевого или рубидиевого электродов следует считать неизменным в различных растворителях, т, е. считать, что э. д. с. Pt(H2)|H+||Cs+|Сs при переходе от одного растворителя к другому изменяется не за счет цезиевого электрода, а только за счет водородного электрода.
Однако неизменность разности потенциалов рубидия и цезия не означает, что каждый из этих потенциалов не изменяется при переходе от растворителя к растворителю - они изменяются, но в одинаковой степени.
Этот вывод был сделан на том основании , что изменение потенциала цепи Hg(Cs) | CsCl | AgCl, Ag при переходе от воды к спирту близко к изменению потенциала цепи Pt(H2)|HCl|AgCl, Ag. В этих цепях анионы одинаковы; следовательно, изменения потенциалов водородного и цезиевого электродов (во всяком случае при переходе от воды к спиртам) близки между собой. Поэтому не было оснований предполагать,, что изменение потенциала цепи Pt(H2)|H+|Сs+| Cs во всех растворителях обязано только водородному электроду; изменение потенциала обязано и водородному и цезиевому электродам. Это говорит о том, что в общем нельзя основывать оценку кислотности в неводных растворах на предположении Плескова.
Предположение Плескова оправдывается но отношению к растворителям с высокой диэлектрической проницаемостью и резко отличной от воды основностью (аммиак и муравьиная кислота), однако нельзя распространить этот результат на другие растворители без экспериментальной проверки.
Строго, единая кислотность, которую мы обозначаем рА, отнесенная к воде в качестве единого стандартного состояния, определяется величиной отрицательного логарифма активности иона МН+:
![]() (2.5.1)
(2.5.1)
где ![]() абсолютная активность иона МН+ ,отнесенная к активности протона в разбавленном водном растворе в качестве единого стандартного состояния.
абсолютная активность иона МН+ ,отнесенная к активности протона в разбавленном водном растворе в качестве единого стандартного состояния.
Такая оценка кислотности является термодинамически строго обоснованной. Единая активность ионов лиония, отнесенная к воде в качестве стандартного состояния, может быть выражена так:
![]() (2.5.2)
(2.5.2)
Подставляя эту величину в уравнение (2.5.1), получим:
![]() (2.5.3)
(2.5.3)
в котором активность а* отнесена к бесконечно разбавленному раствору ионов в неводной среде, а коэффициент ![]()
![]() отнесен к воде в качестве стандартного состояния.
отнесен к воде в качестве стандартного состояния.
Величина —lg а*МН+ называется рНр. Она может быть измерена для любого неводного раствора против стандарта в том же самом неводном растворе. В определении этой величины затруднений нет.
Следовательно
pA = pHр![]() (2.5.4)
(2.5.4)
Это однозначное определение величины рА.
2.6. Применение средних коэффициентов активности ![]() ионов для оценки единой шкалы кислотности
ионов для оценки единой шкалы кислотности
Для оценки единой шкалы кислотности можно воспользоваться средними коэффициентами активности ионов сильной соляной кислоты.
Было установлено, что они могут быть определены только для суммы электролитов в целом. Эти величины хорошо известны для ионов ряда сильных кислот, особенно для HCl во многих растворителях. Например, для этилового спирта 2lg![]() . Однако какая часть величины 5,02 составляет lg
. Однако какая часть величины 5,02 составляет lg ![]() и какая часть lg
и какая часть lg![]() мы не знаем. В связи с этим было предложено поступать так: принять, что средний коэффициент активности ионов кислоты равен коэффициенту активности ионов лиония, т. е. предположить, что:
мы не знаем. В связи с этим было предложено поступать так: принять, что средний коэффициент активности ионов кислоты равен коэффициенту активности ионов лиония, т. е. предположить, что:
lg![]() (2.6.1)
(2.6.1)
Из этого предположения следует, что величина рА определяется выражением
![]() (2.6.2)
(2.6.2)
Такой прием вносит определенную ошибку, обусловленную различием в величинах ![]() протонов и анионов кислот, однако сравнение
протонов и анионов кислот, однако сравнение ![]() для разных кислот показало, что эти величины близки между собой; например,
для разных кислот показало, что эти величины близки между собой; например,
и этиловом спирте ![]() ,
, ![]() ,
, ![]() и т.д. Это же наблюдается и для других растворителей.
и т.д. Это же наблюдается и для других растворителей.
Эксперименты показывают, что величины ![]() ионов кислот в спиртам лишь несколько отличаются от величины:
ионов кислот в спиртам лишь несколько отличаются от величины: ![]() ионов солей. Поэтому нельзя предполагать, что изменения энергии сольватации ионов лиония резко отличается от изменения энергии сольватации остальных катионов. Во всяком случае, величина
ионов солей. Поэтому нельзя предполагать, что изменения энергии сольватации ионов лиония резко отличается от изменения энергии сольватации остальных катионов. Во всяком случае, величина ![]() ионов кислот является вполне однозначной и, вероятно, оценивает изменение кислотности с большей надежностью, чем величины Н0, Н(-), рННас и т.д.
ионов кислот является вполне однозначной и, вероятно, оценивает изменение кислотности с большей надежностью, чем величины Н0, Н(-), рННас и т.д.
2.7 Нахождение единой кислотности рА с помощью ![]() протонов
протонов
Все перечисленные выше методы не позволяют однозначно оценить кислотность неводных растворов в единой шкале. Вопрос об этой шкале может быть решен только на основании данных о величинах химической энергии сольватации протонов в различных растворителях. Эти данные получены на основании подсчетов сумм и разностей химических энергий сольватации ионов в неводных растворах из данных об электродвижущих силах цепей без переноса и с переносом в неводных растворах. Путем экстраполяции величин суммарной энергии сольватации ионов водорода и ионов галогенов (ионы галогеноводородных кислот) и разностей энергий сольватации ионов водорода и ионов щелочных металлов была определена энергия сольватации протона и других ионов в различных растворителях.
При переходе от водного к неводному раствору следует считаться с том, что протяженность шкалы различна для разных растворителей. Для того чтобы оценить абсолютную кислотность, кроме протяженности шкалы нужно знать, как смещено начало шкалы кислотности одного растворителя но отношению к шкале кислотности воды.
Использование ![]() протонов в различных растворителях в качестве единой меры изменения кислотности в разных растворителях однозначно характеризует величину смещения шкал кислотности.
протонов в различных растворителях в качестве единой меры изменения кислотности в разных растворителях однозначно характеризует величину смещения шкал кислотности.
Обозначим начало шкалы для воды через 0; шкала для этилового спирта имеет протяженность 19,3; если ![]() = 4,2, то очевидно, что шкала в этиловом спирте начинается в области —4,2 и заканчивается при рА = 15,1. У метилового спирта
= 4,2, то очевидно, что шкала в этиловом спирте начинается в области —4,2 и заканчивается при рА = 15,1. У метилового спирта ![]() = 3,3, а вся шкала 16,9; шкала для него расположится от - 3,3 до +13,6;
= 3,3, а вся шкала 16,9; шкала для него расположится от - 3,3 до +13,6; ![]() в муравьиной кислоте
в муравьиной кислоте ![]() = 8,6, вся шкала равна 6,1, она расположена меж
= 8,6, вся шкала равна 6,1, она расположена меж